
Theoretical calculations for electronic elucidation of rotational barriers: theory and applications
DOI:
https://doi.org/10.20873/jbb.uft.cemaf.v8n2.bihainKeywords:
theoretical calculations, stereoeletronic effects, EDA, QTAIM, NBOAbstract
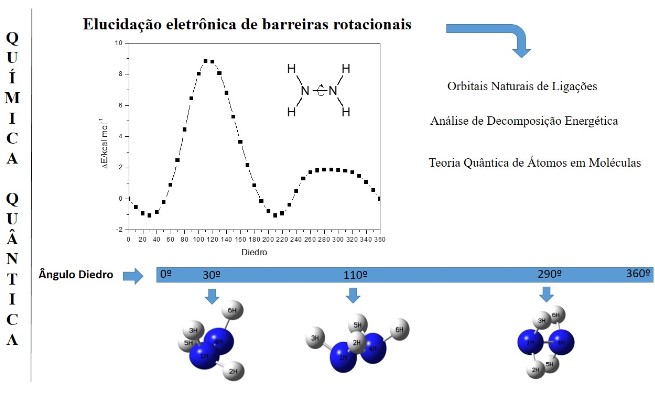
Theoretical calculations of electronic structure are tools of theoretical chemistry. Its importance extends from rotation analyses, hyperconjugation, steric effects on orbital location calculations, energy level and interaction between ligand and antiligantian orbitals. Theories like Natural Bond Orbital (NBO), Quantum Theory of Atoms in Molecules (QTAIM), e Energetic Decomposition Analysis (EDA), are examples of theoretical tools that describe the electronic structure of molecules and solids. In this context, the aim of the article was to describe the theoretical approaches of NBO, EDA and QTAIM, briefly with its applications in the chemical system hydrazine and in theoretical works found in the literature that reinforce the elucidation off stereoelectronic effects.
References
Anderson JSM. et al. Molecular QTAIM Topology Is Sensi-tive to Relativistic Corrections. Chemistry - A European Journal, v.25, n.10, p.2538-2544, 2019.
https://doi.org/10.1002/chem.201804464
Arputharaj DS. et al. Topological Electron Density Analysis and Electrostatic Properties of Aspirin: An Experimental and Theoretical Study. Crystal Growth & Design, v.12, n.9, p.4357-4366, 2012.
https://doi.org/10.1021/cg300269n
Bader RFW. Atoms in molecules. Accounts of Chemical Research, v.18, n.1, p.9-15, 1985.
Bader RFW, Nguyen-Dang TT. Quantum Theory of Atoms in Molecules-Dalton Revisited. In: LÖWDIN, P.-O. Advances in Quantum Chemistry. Academic Press, v.14, p.63-124, 1981.
Clauss AD, Nelsen SF, Ayoub M, Moore JW, Landis CR, Weinhold F. "Rabbit Ears Hybrids, VSEPR Sterics, and Other Orbital Anachronisms", Chem. Educ. Res. Pract., v.15, p.417-434, 2014.
Cukrowski, I. IQA-embedded fragment attributed molecular system energy change in exploring intramolecular interac-tions. Computational and Theoretical Chemistry, v.1066, p.62-75, 2015.
https://doi.org/10.1016/j.comptc.2015.04.018
De Aguiar Filho SQ. et al. Theoretical study of the internal rotational barriers of fluorine, chlorine, bromine, and iodine-substituted ethanes. Computational and Theoretical Chemis-try, v.1166, p.112589, 2019.
https://doi.org/10.1016/j.comptc.2019.112589
Dennington R, Keith T, Millam J. (2009) Gauss View, Ver-sion 5. Semichem Inc., Shawnee Mission.
Dreizler RM, Gross EKU. Density Functional Theory: An Approach to the Quantum Many-Body Problem. Springer-Verlag, New York, 1990.
Foster JP, Weinhold F. Natural hybrid orbitals. Journal of the American Chemical Society, v.102, n.24, p.7211-7218, 1980.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, LI X, Hratchian, HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Ha-da M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, NakaI H, Vreven T, Montgomery JA, Peralta JR, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gompert R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, REVISION D.01, GAUSSIAN, INC.: WALLINGFORD CT, 2009.
Gangadharan P, Krishnan S. Physica polonica a natural bond orbital (NBO) Population Analysis of 1-Azanapthalene-8-ol Rubarani, v.121, n.1, 2013.
https://doi.org/10.12693/APhysPolA.125.18
Glendening ED, Landis CR, Weinhold F. Natural bond orbital methods. WIREs Computational Molecular Science, v.2, n.1, p.1-42, 2012.
Grabowski SJ. What Is the Covalency of Hydrogen Bonding? Chemical Reviews, v.111, n.4, p.2597-2625, 2011.
https://doi.org/10.1021/cr800346f
Hathwar VR. et al. Charge Density Analysis of a Pentaborate Ion in an Ammonium Borate: Toward the Understanding of Topological Features in Borate Minerals. The Journal of Physical Chemistry A, v.115, n.45, p.12818–12825, 2011.
https://doi.org/10.1021/jp203983v
Hehre WJ. Ab initio molecular orbital theory. Accounts of Chemical Research, v.9, n.11, p.399–406, 1976.
https://doi.org/10.1021/ar50107a003
Juaristi E, Cuevas G. Recent studies of the anomeric effect. Tetrahedron, v.48, n.24, p.5019–5087, 1992.
Khaliullin RZ. et al. Unravelling the Origin of Intermolecular Interactions Using Absolutely Localized Molecular Orbitals. The Journal of Physical Chemistry A, v.111, n.36, p.8753–8765, 2007.
Kumar PSV, Raghavendra V, Subramanian V. Bader’s Theo-ry of Atoms in Molecules (AIM) and its Applications to Chemical Bonding. Journal of Chemical Sciences, v.128, n.10, p.1527–1536, 2016.
https://doi.org/10.1007/s12039-016-1172-3
Landis CR, Weinhold F. "The NBO View of Chemical Bond-ing", in, G. Frenking and S. Shaik (eds.), The Chemical Bond: Fundamental Aspects of Chemical Bonding, p. 91-120, 2014.
Lapointe SM. et al. QTAIM study of an alpha-helix hydrogen bond network. The Journal of Physical Chemistry. B, v.113, n.31, p.10957-10964, 2009.
https://doi.org/10.1021/jp903635h
Liu S. Origin and Nature of Bond Rotation Barriers: A Uni-fied View. The Journal of Physical Chemistry A, v.117, n.5, p.962-965, 2013.
https://doi.org/10.1021/jp312521z
Liu S. Steric effect: A quantitative description from density functional theory. The Journal of Chemical Physics, v.126, n.24, p.244103, 2007.
https://doi.org/10.1063/1.2747247
Liu S, Govind N, Pedersen LG. Exploring the origin of the internal rotational barrier for molecules with one rotatable dihedral angle. The Journal of Chemical Physics, v.129, n.9, p.094104, 2008.
https://doi.org/10.1063/1.2976767
Liu S, Rong C, Lu T. Electronic forces as descriptors of nu-cleophilic and electrophilic regioselectivity and stereoselec-tivity. Physical Chemistry Chemical Physics, v.19, n.2, p.1496-1503, 2017.
https://doi.org/10.1039/C6CP06376D
Liu S, Schauer CK. Origin of molecular conformational stabil-ity: Perspectives from molecular orbital interactions and density functional reactivity theory. The Journal of Chemical Physics, v.142, n.5, p.054107, 2015.
https://doi.org/10.1063/1.4907365
Löwdin PO. Quantum Theory of Many-Particle Systems. I. Physical Interpretations by Means of Density Matrices, Natural Spin-Orbitals, and Convergence Problems in the Method of Configurational Interaction. Physical Review, v.97, n.6, p.1474-1489, 1955.
https://doi.org/10.1103/PhysRev.97.1474
Mitoraj MP, Michalak A, Ziegler TA. Combined Charge and Energy Decomposition Scheme for Bond Analysis. Journal of Chemical Theory and Computation, v.5, n.4, p.962-975, 2009.
https://doi.org/10.1021/ct800503d
Møller CHR, Plesset MS. Note on an Approximation Treat-ment for Many-Electron Systems. Physical Review, v.46, n.7, p.618–622, 1934.
Mitoraj M, Michalak A. Natural orbitals for chemical valence as descriptors of chemical bonding in transition metal com-plexes. Journal of Molecular Modeling, v.13, n.2, p.347–355, 2007.
https://doi.org/10.1007/s00894-006-0149-4
Morgon NH, Custodio R. Teoria do Funcional de Densidade. Química Nova (impress), v.18, n.1, p.44-55, 1995
Morokuma, K. Molecular Orbital Studies of Hydrogen Bonds. III. C=O···H–O Hydrogen Bond in H2CO···H2O and H2CO·2H2O. The Journal of Chemical Physics, v.55, n.3, p.1236-1244, 1971.
Mulliken RS, Rieke CA, Brown WG. Hyperconjugation*. Journal of the American Chemical Society, v.63, n.1, p.41-56, 1941.
Oliveira BG, Araújo RCMU, Ramos MN. A topologia mole-cular QTAIM e a descrição mecânico-quântica de ligações de hidrogênio e ligações de di-hidrogênio. Química Nova, v.33, n.5, p.1155-1162, 2010.
https://doi.org/10.1590/S0100-40422010000500029
Parr RG, Yang W. Density-Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989.
Parthasarathi R. et al. Bader’s and Reactivity Descriptors’ Analysis of DNA Base Pairs. The Journal of Physical Chemistry A, v.108, n.17, p.3817-3828, 2004.
https://doi.org/10.1021/jp031285f
Parthasarathi R, Subramanian V. Stacking Interactions in Benzene and Cytosine Dimers: From Molecular Electron Density Perspective. Structural Chemistry, v.16, n.3, p.243-255, 2005.
Parthasarathi R, Subramanian V, Sathyamurthy N. Hydrogen Bonding in Phenol, Water, and Phenol−Water Clusters. The Journal of Physical Chemistry A, v.109, n.5, p.843-850, 2005.
https://doi.org/10.1021/jp046499r
Pavan MS. et al. Characterization of Interactions Involving Bromine in 2,2-Dibromo-2,3-dihydroinden-1-one via Ex-perimental Charge Density Analysis. Crystal Growth & De-sign, v.14, n.11, p.5477–5485, 2014.
https://doi.org/10.1021/cg500659c
Pereira DH. et al. A study of the rotational barriers for some organic compounds using the G3 and G3CEP theories. Journal of Molecular Modeling, v.20, n.4, p.2199, 2014.
Popelier PLA. Quantum Chemical Topology: on Bonds and Potentials. In: WALES, D. J. (Ed.). Intermolecular Forces and Clusters I. Berlin, Heidelberg: Springer Berlin Heidel-berg, v.115, p.1–56, 2005.
Popelier PLA, Bader RFW. The existence of an intramolecular C-H-O hydrogen bond in creatine and carbamoyl sarcosine. Chemical Physics Letters, v.189, n.6, p.542-548, 1992.
Raissi, H. et al. Intramolecular Hydrogen Bonding in Structur-al Conformers of 2-Amino Methylene Malonaldehyde: AIM and NBO Studies. International Journal of Quantum Chem-istry, v.110, p.821-830, 2009.
https://doi.org/10.1002/qua.21795
Reis D, Ribeiro I, Pereira DH. DFT study of the application of polymers cellulose and cellulose acetate for adsorption of metal ions (Cd2+, Cu2+ and Cr3+) potentially toxic. Poly-mer Bulletin, 2019.
Su P, Li H. Energy decomposition analysis of covalent bonds and intermolecular interactions. The Journal of Chemical Physics, v.131, n.1, p.014102, 2009.
https://doi.org/10.1063/1.3159673
Thakur TS, Desiraju GR. Theoretical investigation of C–H⋯M interactions in organometallic complexes: A natural bond orbital (NBO) study. Journal of Molecular Structure: THEOCHEM, v.810, n.1, p.143-154, 2007.
https://doi.org/10.1016/j.theochem.2007.02.012
Todd A, Keith Aimall (Version 17.11.14), TK Gristmill Soft-ware, Overland Park KS, USA, 2017 (aim.tkgristmill.com)
Tsuboi M, Overend J. Amino wagging and inversion in hy-drazines: RR branch of the antisymmetric wagging band of NH2NH2 J Mol Spectrosc, v.52, p.256-268, 1974.
Vener MV. et al. QTAIM Study of Strong H-Bonds with the O−H···A Fragment (A = O, N) in Three-Dimensional Peri-odical Crystals. The Journal of Physical Chemistry A, v.111, n.6, p.1155-1162, 2007.
https://doi.org/10.1021/jp067057d
Vorobyov I, Yappert MC, Dupré DB. Energetic and Topolog-ical Analyses of Cooperative σH- and πH-Bonding Interac-tions. The Journal of Physical Chemistry A, v.106, n.44, p.10691-10699, 2002.
https://doi.org/10.1021/jp0264580
Weinhold FR, Klein R. "What is a Hydrogen Bond? Reso-nance Covalency in the Supramolecular Domain", Chem. Educ. Res. Pract. v.15, p.276-285, 2014.
https://doi.org/10.1039/C4RP00030G
Weinhold F. Natural bond orbital analysis: A critical overview of relationships to alternative bonding perspectives. Journal of Computational Chemistry, v.33, n.30, p.2363-2379, 2012.
https://doi.org/10.1002/jcc.23060
Zhao L. et al. Energy decomposition analysis. WIREs Compu-tational Molecular Science, v.8, n.3, p.e1345, 2018.
Zhou X. et al. Anomeric effect revisited: Perspective from information-theoretic approach in density functional reactivi-ty theory. Chemical Physics Letters, v.684, p.97-102, 2017.
Zhurov VV, Pinkerton AA. Inter- and Intramolecular Interac-tions in Crystalline 2-Nitrobenzoic Acid—An Experimental and Theoretical QTAIM Analysis. The Journal of Physical Chemistry A, v.119, n.52, p.13092-13100, 2015.
https://doi.org/10.1021/acs.jpca.5b10027
Ziegler T, Rauk A. On the calculation of bonding energies by the Hartree Fock Slater method. Theoretica chimica acta, v.46, n.1, p.1-10, 1977.
Published
How to Cite
Issue
Section
License
Copyright (c) 2024 - Journal of Biotechnology and Biodiversity

This work is licensed under a Creative Commons Attribution 4.0 International License.
Authors who publish with this journal agree to the following terms:
Authors retain copyright and grant the journal right of first publication with the work simultaneously licensed under a Creative Commons Attribution License (CC BY 4.0 at http://creativecommons.org/licenses/by/4.0/) that allows others to share the work with an acknowledgement of the work's authorship and initial publication in this journal.
Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the journal's published version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgement of its initial publication in this journal.
Authors are permitted and encouraged to post their work online (e.g. in institutional repositories or on their website) prior to and during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (Available at The Effect of Open Access, at http://opcit.eprints.org/oacitation-biblio.html).
